TL:DR Executive Summary:
- The size of a “small segment” is not strictly defined but is generally less than 10 cM and preferably less than 7 cM;
- A very large percentage of small segments are not valid matching segments (i.e., are false segments), and our ability to distinguish valid segments from false segments is either impossible or very limited;
- Although we cannot reliability utilize a small shared segment as genealogical evidence in most cases, that does not negate the potential value of a genealogical relationship;
- There is no evidence that: (1) triangulating a small segment; or (2) sharing a large segment in addition to the small segment; or (3) finding a shared ancestor, increases the probability that a small segment is valid;
- Even if a small segment is somehow identified as valid, perhaps an even bigger challenge is determining whether the small segment (which can be 10, 20 or more generations old) came from the identified ancestral line or another known or unknown shared ancestral line.
Small segments have long been a controversial subject in the genealogy community. Some love them, some hate them. Here we will look at all the available evidence surrounding small segments, some of the misconceptions associated with them, and some of the ways we might be able to utilize small segments in our research.
What are “Small Segments”?
One of the biggest problems surrounding the discussion of small segments it that there is no strict definition of the term. A “small segment” to one person might be anything 5 cM or less, while a “small segment” to another person might be anything less than 10 cM. And there are many other variations. For purposes of this analysis, we will define a small segment as any single segment of DNA less than 8 cM (although segments as high as 10 cM or more can be problematic as well).
There is no question among geneticists (and most experienced genetic genealogists) that small segments are notoriously problematic. ALL available evidence suggests that a significant percentage (ranging from almost every segment to most segments to some segments, depending on the cM threshold) of small segments are not actually IBD (“identical by descent”), meaning they are not actually shared by the two individuals. Rather, the segment is a false match due artificial matching between the two individuals.
What Causes False Matching?
False matching is typically due to the existence of a “pseudosegment.” A pseudosegment is a stretch of DNA that artificially weaves back and forth between the maternal copy of the chromosome and the paternal copy of the chromosome to create a stretch of DNA that doesn’t actually exist in the individual. These pseudosegments are created as an artifact of microarray testing, which is the form of testing all of the genetic genealogy testing companies currently use. Since the results aren’t oriented by “maternal copy” and “paternal copy” of the DNA, the matching algorithm must be allowed to weave back and forth between the DNA results in order to check for matching. Most of the time, the weaving recreates either the “maternal copy” or the “paternal copy.” However, the smaller the cM threshold, the more common it is to weave back and forth among copies (which doesn’t really happen!).
For more about pseudosegments, see: “A Small Segment Round-Up.”
What Evidence is Available?
All of the genetic genealogy testing companies, with the exception of MyHeritage, have published data emphasizing the danger of small segments. Below, we will examine all this evidence in turn.
1. 23andMe
In general, 23andMe has a minimum segment threshold of 7 cM for matching, meaning that two people must share at least a 7 cM segment to be identified as a match.
In 2014, the geneticists at 23andMe published a peer-reviewed paper about false segments. See “Reducing Pervasive False-Positive Identical-by-Descent Segments Detected by Large-Scale Pedigree Analysis” Molecular Biology and Evolution, Volume 31, Issue 8, August 2014, Pages 2212–2222.
To identify false segments, the researchers looked at matching between tested triads (mother, father, child) and other people in the database. If a segment matched a child and person X, AND that same segment matched the child’s parent and person X, then the segment was identified as a valid segment. If a segment matched a child and person X, and did NOT match either of the child’s parent, then the segment was identified as a false segment. They did this for a range of segments from about 2 cM up to 6 cM.
As shown in Supplemental FIG. S5, many small segments (the white region) were false. At 2 cM, approximately 70% of segments were false; at 6 cM, approximately 15% of segments were false.
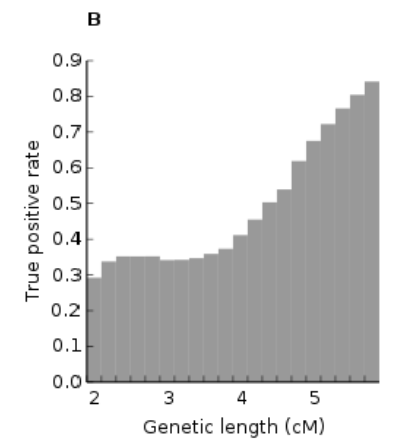
Thus, the research from 23andMe shows that many small segments are false. While having a parent tested clearly helped eliminate many false segments, the problem is likely worse than this research shows. Specifically, while comparing the child’s segments to the parent’s segments helped eliminate matching on the CHILD’S side, there does not appear to have been any such elimination on the other side of the match.
2. AncestryDNA
As of the summer of 2020, AncestryDNA’s matching threshold is 8 cM, meaning that two people must share at least one 8 cM segment to be identified as a match. Prior to the summer of 2021, AncestryDNA provided matches sharing as little as 6 cM. Although the primary reason for eliminating segments less than 8 cM was likely due to storage constraints, Ancestry also recognized that a significant percentage of segments less than 8 cM are false segments, and our ability to distinguish between false and valid segments is limited.
The AncestryDNA Matching White Paper (July 15, 2020) describes in detail the process for identifying matches within the AncestryDNA database. The paper also reports on an experiment similar to the experiment from 23andMe’s 2014 paper. In FIG. 3.3, the researches look at concordance by identifying the frequency at which a child shares IBD (i.e., shared DNA) with one or both parents, from 6 cM to 20 cM. The black shading in each bar is the frequency of IBD that a child shares with a match but does NOT share with either parent (and is thus false matching). At 6 cM, it is 50%. At 7 cM it is approximately 30%. At 10 cM, it is roughly 15%. This clearly demonstrates that the matching algorithm is identifying a large number of false matches for segments less than 10 cM (with the problem growing as the segments get smaller).
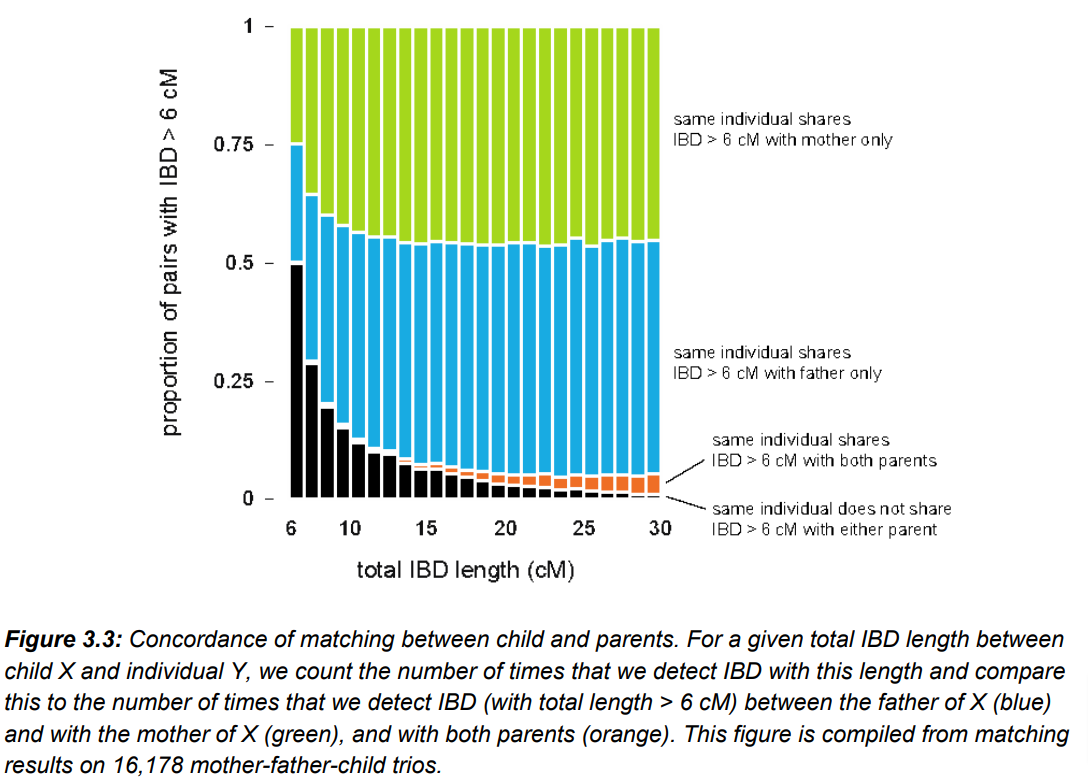
As with the research from 23andMe, the data from AncestryDNA suggests that many small segments are false.
3. The Family Tree DNA
Family Tree DNA was once a proponent of small segments, and even provided very small segment data (as low as 1 cM), but in the summer of 2021 they made drastic changes to their matching algorithm. Most relevant to the current topic, they stopped identifying shared segments of DNA less than 6 cM. Although for several reasons this change didn’t receive as much fanfare or backlash as the AncestryDNA change, in many ways it was even more significant.
In the new white paper associated with the changes to the matching algorithm (“Family Finder Matching 5.0: Matching Algorithm and Relationship Estimation”), FTDNA created a simulated dataset of matching segments. They randomly selected 4,000 unrelated samples as founders from Western European and Ashkenazi Jewish populations to represent nonendogamous and endogamous populations, respectively. The simulated relationship ranged from parent/child and full siblings up to 6th cousins as shown in the following pedigree.
FTDNA used the simulated dataset to test valid (IBD) versus false segments. In the simulated dataset, we know which segment is inherited from which ancestor, so those segments can be used as ground truth data to compare with the detected segments for validation purposes. The result of this analysis is shown in Table 2 of the white paper.
At 6 cM, the percentage of false segments (“False positive rate”) was about 20%, meaning one out of every five segment of 6 cM was a false segment. Below 6 cM the false positive rate skyrocketed, and above 6 cM the false positive rate dropped dramatically. Following this analysis, FTDNA chose “6 cM as a reasonable threshold to report IBD segments.” Notably, however, FTDNA acknowledges that many segments of 6 and even 7 cM are false, and does not identify a method for their matching algorithm (or any other method) to distinguish between valid and invalid segments.
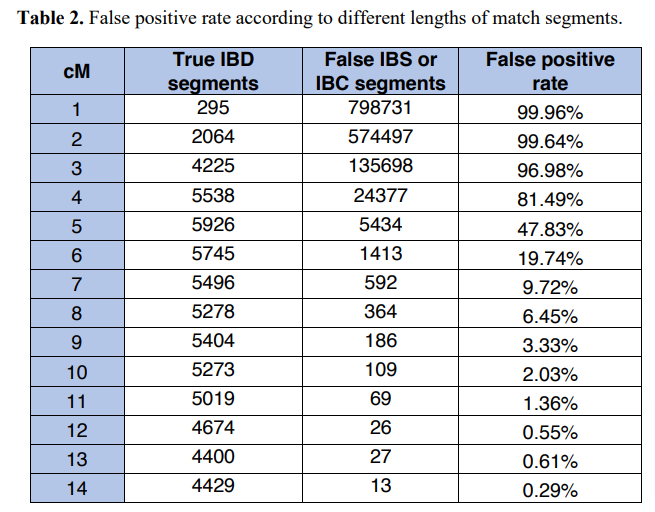
With this research, FTDNA joins 23andMe and AncestryDNA in the determination that many small segments are false.
4. Anecdotal Evidence
In addition to the peer-reviewed or large dataset analyses described above, there is some anecdotal evidence to suggest that many small segments are false.
For example, in 2017 I compared my match list at AncestryDNA to the match list of my tested parents in order to identify how many of my matches were not shared by either parent. This is, of course, biologically impossible and any such “unshared” matches must be false positives or false negatives. In many or most cases, they are likely to be false matches resulting from small false segments. Note that this was before the elimination of segments/matches below 8 cM.
I found that approximately 32% of my 16,193 matches were not shared with either parent. Of those, 60% were less than 7 cM, and 95% were less than 10 cM. See “The Danger of Distant Matches.” Once again, this suggests that many small segments/matches are false segments/matches.
This analysis was repeated by others, including Debbie Kennett (see “Comparing parent and child matches at AncestryDNA” and “Comparing match tallies for family members with Family Tree DNA’s Family Finder test”). Debbie found that 36% of her matches at AncestryDNA were not shared with either parent, and the vast majority of those matches shared less than 10 cM with her.
Several others have repeated this analysis and found similar results (see the links in Debbie’s blog posts).
Thus, this anecdotal evidence repeated by numerous genetic genealogists suggests that small segments/matches are overwhelmingly false and problematic.
Small Segment Misconceptions
Over the years, many small segment misconceptions have gained ground. These misconceptions are usually grounded in confirmation bias, or an attempt to find ways to utilize small segments despite the difficulty in identifying valid versus false segments. Below we will briefly examine some of these misconceptions.
- Triangulation Can Distinguish Between Valid and False Segments
There is no evidence that triangulation increases the likelihood that a triangulated segment is valid. While one “leg” of the triangulation group might be valid, or even two “legs” might be valid, there is no guarantee that the other leg or legs are valid.
We often see this misconception applied as a dual misconception. First, a genealogist will assert that the small segment triangulates and thus is valid. Second, the genealogist will often assert that two of the legs of the triangulation group are closely related (parent/child, siblings, first cousins, etc.) which of course cannot form different legs of the triangulation. Rather, close relatives form a single leg of the triangulation group since they would have inherited the segment from the same very recent ancestor (a parent, grandparent, etc.).
While many people triangulating on a small segment could conceivably suggest that the small segment is valid, ironically that also suggests that the segment is very, very old and therefore not useful other than to perhaps establish a general community connection.
Unfortunately, this misconception is very prevalent.
- Sharing a Large Segment (or Many Small Segments) with a Match Validates a Shared Small Segment
There is similarly no evidence that sharing a large segment in addition to a small segment, or sharing numerous segments, increases the likelihood that a small segment is valid. I did a very small, anecdotal study here: “Small Matching Segments – Friend or Foe?”
- A SNP-Dense Small Segment is Likely to be Valid
Unfortunately, there is no evidence that sharing a SNP-dense small segment (meaning there are many tested SNPs in the segment) increases the likelihood that a small segment is valid. Although conceptually this seems like a very logical hypothesis, there is currently no evidence to support the conclusion.
- Finding a Genealogical Connection Validates a Shared Small Segment
This is probably the most common, and most appealing, misconception. It is tightly intertwined with confirmation bias.
There is no evidence that identifying a shared ancestor increases the likelihood that a small segment shared with that match is valid. One does not go hand-in-hand with the other. For example, I can share very recent common ancestry with Person Y and NOT share any DNA with them; even as recent as a shared great-great-grandparent. Sharing common ancestry and sharing DNA are not synonymous.
I have 1000s of matches below 10 cM. How could I not share a common ancestor with at least some of those matches? If I picked 500 random people in the AncestryDNA database with whom you do NOT share any DNA (which is essentially what false matching does!), and I asked you to find a common ancestor with some of these matches, of course you would be able to do that. It would be impossible for you not to be able to do it! There were only so many people on earth, after all!
Even assuming, for the sake of argument, that sharing a common ancestor and sharing a small segment could mean that the small segment is – or is more likely to be – valid, there is the perhaps even more significant issue of age of the segment. A 7 cM segment, for example, can be 10 or 20 generations old. No one has a complete tree out to 20 generations (and, if they do, it is inaccurate due to unknown Misattributed Parentage Events (MPEs)), and thus the ability to determine what ancestors they do and do not share at that vast generational distance is impossible.
- Phasing Eliminates Small Segments and False Matching
Phasing is the separation of a test-taker’s DNA into the paternal copy of the chromosome, and the maternal copy of the chromosome. Phasing can be done in at least three different ways: (1) by direct comparison to one or both parents; (2) by aligning thousands of matches along your chromosomes (using AncestryDNA’s SideView technology, for example); and (3) using population genetics and large datasets of DNA haplotypes. Several of the testing companies using method #3 (population genetics) to phase DNA before matching; despite this, we know there continue to be false segments/matches.
Theoretically, when DNA is phased, the pseudosegments that result in false matching can no longer be created (because the matching algorithm cannot artificially weave back and forth between the maternal and paternal DNA). However, while phasing is likely to eliminate many or most false small segments, it cannot eliminate all false matches.
Further, while I might be able to phase my DNA (at GEDmatch, for example), this does NOT phase the DNA of all of my matches. Accordingly, while I may have a valid small segment as a result of phasing (again, not something I can definitely prove), my match may only share this DNA because it is a pseudosegment for them.
For more about phasing and false matching, see “GUEST POST – What a Difference a Phase Makes” and “The Effect of Phasing on Reducing False Distant Matches (Or, Phasing a Parent Using GEDmatch).”
Small Segment Caveats
It is very important to note that although a small segment cannot reliably be utilized as genetic evidence for a particular relationship, that does not negate the identified genealogical relationship! For example, in the AncestryDNA ThruLine presented below, I share a single 8 cM segment with a 4C1R (fourth cousin once removed). If I were going to use this 8 cM segment as genetic evidence, I would need to definitely resolve two issues: (1) is the segment a real segment?; and (2) is the segment (which can be very old) from this line rather than another shared line? As we know, finding a genealogical connection does NOT validate this segment in any way at all. Neither does triangulating. So we have a huge uphill battle in order to utilize the segment as genetic evidence.
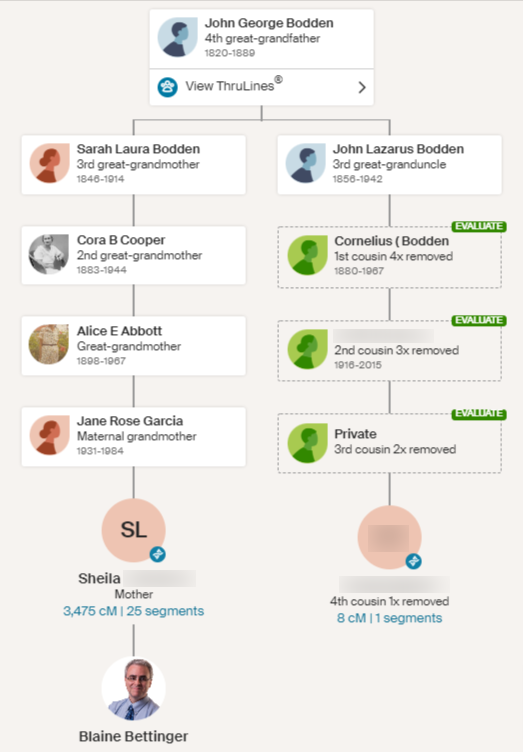
HOWEVER! Identifying this genealogical relationship is still valuable! I can contact this person, I can work with her to collaborate on this line, and so on. Maybe they have records and photographs I don’t have, for example. Do not throw out a genealogical relationship just because the connection is identified via a small segment.
BUT! Do not make the similar mistake of utilizing the small segment as evidence of the relationship, just because you’ve found a genealogical connection. The two do NOT go hand in hand.
Small Segment Uses?
With all this in mind, are there ever circumstances in which a small segment could reliably be identified as valid and as useful genetic evidence?
1. VERY Close Relative Comparisons
The most common scenario I’ve found where it is relatively easy to identify a small segment as valid is by comparing a grandchild to a grandparent, either directly or indirectly (i.e., via visual phasing). The ends of the chromosomes are recombination hotspots, and it is VERY common for small segments to be formed at the end of the chromosomes. In the following, for example, there is a 4 cM segment at the end of chromosome 16 that is from my paternal grandmother:
These small segments will only form at the ends of the chromosomes. Because of the phenomenon of recombination interference, these small segments do not form in the interior of the chromosome. Thus, I would discount a small segment I might identify in the middle of a chromosome when comparing a tested grandparent to a tested grandchild, for example. Since I can follow these small segments via recombination, they are almost certainly valid segments.
However, if I were to compare another close relationship such as aunt/niece and identified a small segment, regardless of where it were along the chromosome, I would be far less likely to believe it were valid, or to utilize it any way.
2. An Overlapping Large Segment
This is perhaps the most common defense of small segments, and it is unfortunately a bit of a strawman argument. Essentially, someone will argue that they share (for example) an 8 cM segment with John Doe. But their sibling or uncle or etc. shares a 20 cM segment with John Doe in the same, overlapping region.
This is NOT a defense small segments. What it means, is that the test-taker is working with a large, 20 cM segment (although age of the 20 cM segment can still be an issue). Without the larger overlapping segment, the test-taker would be no closer to being able to utilize the lone small segment. With the large, 20 cM segment, there is absolutely no need to work with the small segment.
This is also yet another great reason to test as many family members as possible!
3. Population Genetics
There is some suggestion that small segments can be used on a large scale to identify older biogeographical origins. For example, many people with African ancestry have found African matches and this may point to biogeographical origins. There is a GREAT blog post from Tracing African Roots about the dangers and potential rewards of this approach: “How to find those elusive African DNA matches on Ancestry.”
Much more research and comparative analysis is needed in this area (especially to avoid the allure of confirmation bias), but there is a great deal of potential!
4. The Future?
In the future, we may be able to more reliably utilize small segments with whole-genome sequencing and very rare SNPs. This could attack both the validity and age questions when it comes to small segments. But it would require both whole-genome sequencing and many, many genomes (to identify rare SNPs), and thus is likely to be far in the future.
Conclusions
Small segments are the Sirens of genealogy. They lure us in with promise, and then lead us to a disastrous end. Everyone is free to use small segments as they wish, but as genealogists and scientists it is our duty to critically analyze evidence and only use it for what it CAN tell us, not what we want it to tell us. In order to utilize a small segment, both issues must be resolved: (1) validity of the segment; and (2) age of the segment. Only then can we reliability utilize a small segment as genetic evidence.
Links For More Information:
- Family Locket – “Is This a False Segment?” (May 7, 2022)
- The DNA Geek – “The Small Segment Debate is Over” (Sept. 6, 2021)
- The Legal Genealogist – “Don’t Poison That Tree” (Sept. 5, 2021)
- Cruwys News – “Comparing parent and child matches at AncestryDNA” (Aug. 6, 2017)
- Cruwys News – “Comparing match tallies for family members with Family Tree DNA’s Family Finder test” (July 29, 2017)
- DNA Sleuth – “When is a match a false positive?” (July 8, 2016)
- Cruwys News – “Tracking DNA segments through time and space” (Apr. 26, 2015)
- Your Genetic Genealogist – “The Folly of Using Small Segments” (Dec. 3, 2014)
.
Many thanks, Blaine, for this excellent summary of the current state of the “small segments issue.”
As a corollary, I believe the matter is exacerbated in certain instances when attempting to compare data between microarray tests/versions that target only a small percentage of the same SNPs, in some cases as few as 18% of the tested loci. I have no information about the extent and effectiveness of genotype imputation for this purposes as used by FTDNA, MyHeritage, or Living DNA–the only major testing companies that accept third-party raw data–but since GEDmatch does not rely on imputation we can see some stark indicators there.
As an example, my publicly-visible “superkit” at GEDmatch is data extracted from a 30X whole genome sequencing and included about 2.1 million SNPs as uploaded, which would represent a dataset almost 3.5 times larger than the typical microarray results. Hypothetically, the more tested SNPs, the better the accuracy should be because, in part, the likelihood should be greater that haplotype switching errors could be detected and used to better determine continuity mismatches within a segment…indicating the segment is not actually a continuous block of DNA.
At GEDmatch, I also have 11 kits flagged as “research” that were constructed using the same WGS data, each using precisely the same SNPs examined by different test versions at different companies. If I run a Tier 1 one-to-many comparison at default settings for all matches >= 10cM–so this is already screening out segment sizes below 10cM–my superkit reports 9,242 matches, and 95.55% of those come from other people’s kits whose results contain greater than 99,000 of the same SNPs as my superkit. When I do the same for the research kit with the greatest disparity of results, I get 24,996 reported matches, and only 12.28% come from kits with which I have 99,000 or more of the same SNPs tested. The accuracy in comparison to the WGS superkit varies by microarray test and version, but at the edge of the spectrum 15,754 out of 24,996 reported matches, fully 63% of them, do not appear when comparing to the superkit.
This, of course, is not direct evidence that all 63% can be attributed to false-positives. But the number of matches begins to coalesce into agreement among the 12 different kits at the level of 20cM minimum, and by 30cM the counts are in 95.9% concordance. The numbers would seem to infer that at GEDmatch, and using default parameters, two kits with low in-common SNP counts might become less and less accurate in the comparisons of small segments as the individual segment sizes decrease below 20cM or even 30cM, and even at 10cM it could be possible that one in two reported matches might be false-positive.
I completely agree that in many cases, while we may not be comparing apples to oranges, we are comparing Macintosh to Granny Smith and it definitely exacerbates false matching. Of course, even if we could reliably determine which segments are valid and which are invalid, we still have the age of the segment challenge to overcome!
Thank you Blaine for all you do. I know this is probably folly, but I took a different approach to compare BigY DNA with small segment triangulations to eliminate useless triangulations. This is important. I have spent a 1000+ hours on a particular surname, and about 10 days on just this aspect of it. Even though I’d seen this pattern over and over, I was surprised with the results.
Main Investigation: Can small cM’s triangulations compared to YDNA and BigY branches be used to discern support for our lineages?
Question: Using 3 cM’s upwards for triangulations combined with YDNA and BigY haplogroups, what percentage of the time will a triangulation occur that cannot explain an IBD connection between cousins for a particular ancestor/ancestral couple?
I compared possible cousins between haplogroup branches that have a previous common haplogroup as well as haplogroup branches that don’t have a common ancestor till abt. 63,000 years ago (E-M35 & R-M269) There are 3 possibilities.
1. Three people triangulate with only one branch of a BigY Haplogroup project.
2. Three people triangulate with a previous common haplogroup (further back in time).
3. Three people triangulate with a haplogroup that doesn’t meet up till about 63,000 years ago.
To do this I:
Used only non-phased kits because of the rarity of phased kits on Gedmatch.
Doublecheck any discrepancies against primary documentation to get each person’s tree back to it’s earliest proven ancestor. I wanted to make sure I was comparing Apples to Apples.
Doublecheck the oldest known ancestor in each triangulation to determine their BigY haplogroup as per FTDNA.
In this first trial, I compared kits on Gedmatch on Chromosome 18. Both the E-M35 and the R-M269 groups had triangulations on chromosome 18 suggesting I might get a triangulation between the two distant haplogroups.
Results:
450+ comparisons made between possible cousin pairs.
6 triangulations between members matching one BigY branch of the FTDNA male YDNA timetree. (66.6%)
0 triangulation between members matching a junction of two BigY haplogroups from FTDNA.
3 triangulations between people from the E-M35 and R-M269 haplogroups. (It could be a triangulation for a different surname in common, a wife’s second husband’s line, or accidental matches.) (Not IBD for the ancestor or ancestral couple studied.)
The percentage of the triangulations that have closer cousins with larger cM matches. 44% were comparing one pair of cousins whom were much closer to each other having 31 cM’s and upwards on chromosome 18.
10+ branches compared (The plus is here because 2 more out of 30 people were from lines whose ancestry I could not determine in a way that verifies how they connect with the other 28 persons. These 28 persons did triangulate is ways that were consistent with BigY and previous triangulations that sorted them into groups. In other words, 100% of the ones that were NOT eliminated by having 2 totally different haplogroups, did explain a consistent branch or haplogroup. Two branches out of 10+ possible branches, had consistent triangulations in this study. No one triangulated for the other 8+ branches on chromosome 18.
Sixty-Six.Six (66.6%) of the triangulations occurred on single branches of the male haplogroup tree. None occurred in this study accounting for the junction point between 2 branches, though I have had that occur in this very extensive surname project study. To repeat: After 33.3% of the total triangulations were eliminated because they were triangulating with people whose BigY’s didn’t have a common YDNA ancestor for 63,000 years, the 66.6% that was left, ALL (100% OF THEM) supported consistent conclusions that backed up 2 different YDNA haplogroup branches.
This study was inspired by a simple question that I asked of FTDNA. Which test is the best DNA test to get to prove our ancestry? Their answer: The truth lies in comparing Autosomal DNA with YDNA. I would go one step further. The truth lies in comparing triangulated autosomal DNA to BigY DNA and YDNA. This study is respectfully submitted as a possible way to corroborate or disprove our lineages.
Summary:
Original Question: Using 3 cM’s upwards for triangulations combined with YDNA and BigY haplogroups, what percentage of the time will a triangulation occur that cannot explain an IBD connection between cousins for a particular ancestor/ancestral couple? Answer for this trial: 33.3%. Sixty-six-point-six percent (66.6%) does match well and only explains a common ancestor/ancestral couple on a particular BigY haplogroup branch.
I’m not a statistician, but doesn’t this suggest a possible avenue for getting reasonable data from triangulations even with small cM’s by eliminating the false matches via BigY groups that are NOT related?
It should be noted that another pattern has occurred possibly by explaining why small cM triangulations of significance occur on Gedmatch. On MyHeritage, I have triangulated at about an average of 9 cM’s with cousins descended from the same people as in Gedmatch where the matches show as 3-5 cM matches. I have had people write me to have me check for triangulations on Gedmatch with cousins that are sharing up to 12 cM’s and on Ancestry. When their DNA is transferred from Ancestry to Gedmatch. They have had the small matches on Gedmatch from 3-5 cM’s instead. We might want to start keeping track of these between sites to possibly verify that the DNA is missing segments when it’s transferred into Gedmatch.
Hi, Blaine, in this so interesting article, you focus on small segments defined as those below 10cms (sort of). Are Ancestry 4th-6th cousin matches regarded then as extremely likely to be valid, and what is the likelihood of having false positives in this group?
I find there are still big problems with some of the matches at Ancestry in the 20 cM to 30 cM range. 28% of my 4th to 6th cousin matches at AncestryDNA do not appear as shared matches with either of my parents. The Shared Matches tool is of course restricted to matches sharing 20 cM or more. For many of these matches there are just small discrepancies which mean that the match doesn’t quite meet the threshold. For example I have one match who shares 21 cM with me but 17 cM with my dad. However, for some of these matches there are major discrepancies. I have one match which shares 28 cM with me but only 9 cM with my mum. I only have one match (a 24 cM match) in the 4th to 6th cousin range which doesn’t match either of my parents.
There is a big problem with false matches at MyHeritage. I have two kits at MyHeritage. One is a transfer and the other is a new test on the Global Screening Array. When I last analysed my matches at MyHeritage I found that 38% of the matches with my transferred kit don’t match either of my parents. With the GSA, 47% didn’t match either of my parents. I think the false matching rate is inevitably going to be much higher with companies that accept transfers because of the small number of overlapping SNPs on the different chips and the need for imputation to fill in the gaps.
In one case I have a 6 cM Ancestry match (from before the cull) with Shared Matches – so 20cM at least to them. And this match is known by conventional genealogy as 3C.
My head tells me this could be an unreliable segment. My heart says the familial connections should count for something. Particularly if the relationship is close and there are lots of matches that also match that segment.
My head comes back and warns me against the slippery slope towards the dark side of a non-scientific approach. So I take that as a one off exception and ignore all the rest.
I’ll just have to look harder at some alternative approaches. Like phasing using siblings’ DNA. And hiring a DeLorean to go back and test my grandparents.
Thanks for all this great information! Such a helpful article.
Thanks for the really helpful article and analysis. You mention above that “Because of the phenomenon of recombination interference, these small segments do not form in the interior of the chromosome.” I was just wondering if this is explained in another post somewhere as I would be really interested in understanding it.
More information and links here: https://en.wikipedia.org/wiki/Crossover_interference
I have to disagree on my endogamous groups Ive ashkenazi and sephardi ancestry all ashkenazi are related indeed so are all European Jews. I match nearly every Moroccan Jew on Gedmatch usually on multiple segments my dad was Moroccan Jewish. Its well known how interrelated Jews are so most segments are real but obviously we cannot find the ancestor usually.
Also a known third cousin shares largest only 12 Cms to say not valid is stupid my mum and her mum grew up together. We know who our ancestors.
I see your point on non Jewish dna Jews are a unique case.